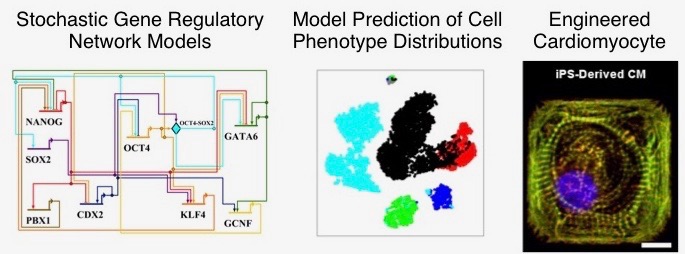
A hallmark feature of many cancers, and an enduring puzzle of biology since its discovery in the 1950’s, is a phenomenon called the Warburg Effect. Here, cancerous cells gain access to oxygen, but then shift their metabolism to aerobic glycolysis, away from (oxygen-using) oxidative phosphorylation (OXPHOS) to favor glycolysis, despite the availability of sufficient levels of oxygen. This is especially intriguing since glycolysis is much less efficient than OXPHOS in producing energy. However, it is increasingly recognized that glycolysis is not a singular choice for all cells in a tumor; OXPHOS modes of metabolism may be dominant to some cells but not others. The proposed outcome of this heterogeneity is that cooperation between these two groups of cancer cells can maximize the delivery and consumption of nutrients and minimize the environmental stresses that are imposed on a tumor. This is known as metabolic symbiosis. For example, glycolytic cells produce lactate that can be used as fuel by cells using OXPHOS. Figure 1 shows metabolism under varying oxygen concentrations.
In this project, students will develop deterministic and stochastic population-based and individual-based mathematical models to determine conditions under which metabolic symbiosis is not just a manifestation of tumor heterogeneity, but is a fundamental aspect of tumor survival. That is, students will determine conditions and parameter ranges for which tumors that display metabolic symbiosis have an enhanced fitness compared to those in which there is metabolic homogeneity. The population-based modeling will use ordinary and stochastic differential equation models while the individual-based modeling will use CompuCell 3D, which is based on the Cellular Potts approach, as the primary modeling tool. We have extensive experience using these approaches. Model predictions will be compared to results in the experimental literature.
The mathematical challenge lies in developing a model that, on the one hand, contains all the relevant biological features needed to compare with experimental data, and, on the other, is simple enough to provide insight into the underlying mechanism of spatial heterogeneity.
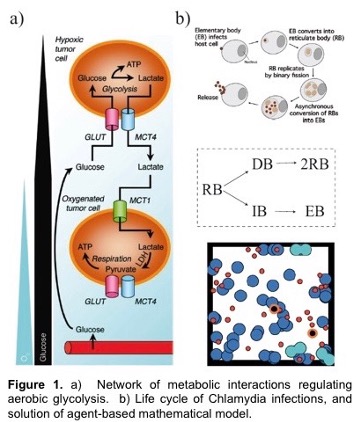
Chlamydia is a bacterium that has a large impact on public health by causing genital, ocular (eye infections) and respiratory diseases. Chlamydia infects a human cell in which it reproduces within a membrane-bound compartment called the chlamydial inclusion. Chlamydia is different from most other infectious bacteria in having an intracellular developmental cycle featuring conversion between two main forms of the bacterium. The reticulate body (RB) is the vegetative but non-infectious form that divides by binary fission. In contrast, the elementary body (EB) cannot divide, and its specialized function is to survive in the extracellular environment and infect new cells (Figure 1b). We would like to define the mechanisms that control the replication of an RB and its conversion into an EB. Our central hypothesis is that RB replication and conversion, and the balance between these processes, are regulated to optimize the production of infectious progeny from an infected cell.
We will develop mathematical models that incorporate the number and location of each chlamydial developmental form, the inclusion size, and the time in the intracellular infection. We will then apply these models to simulate the intracellular infection and compare the results with the data from our 3D reconstructions to identify the critical parameters responsible for the regulation of RB replication and RB-to-EB conversion. The mathematical novelty is in understanding the complex, highly nonlinear optimization process that chlamydia pursues to maximize infection.
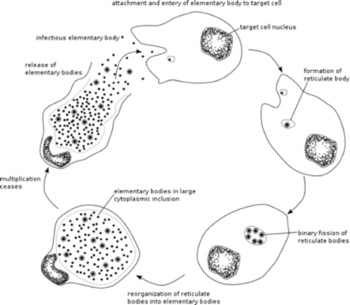
Actin is a major part of the cytoskeletal network influencing cell structure, division, and movement. Understanding the process of actin assembly, where actin monomers (orange circles) are added to a growing actin polymer (orange chain), is therefore critical to understanding these cellular characteristics. Actin assembly is often facilitated by members of a protein family called formins. Formins contain two flexible ‘arms’ (blue chains), which are able to explore the nearby space and bind actin monomers. To facilitate actin assembly, formins perform a ‘capture-and-deliver’, capturing actin monomers in solution and then delivering them to the growing end of the polymer.
The behaviors and mechanisms of different formins are not well understood and are of growing interest in the field. What is it about these flexible arms that allow them to enhance actin polymerization? How do characteristics of specific formins influence this effect? In collaboration with the Quinlan Lab at UCLA, we will investigate how a formin with two flexible ‘arms’ behaves when the arms are tied together at the end (dimerized). Specifically, we are interested in how dimerized formin affects actin assembly compared to monomeric formin. In what way does dimerization influence the ‘capture-and-deliver’ system of formin?
In pursuit of this question, you will introduce dimerization to a pre-existing simulation of formin’s flexible arms. You will then explore how this dimerization impacts the ability of formin to capture and subsequently deliver actin monomers. Skills you will learn include basic biochemistry, polymer physics, reading and understanding experimental data, reading C code, navigating and running scripts in the command line, and writing MATLAB functions to analyze output.
Skin is a richly diverse organ system that requires constant communication between cells and tissues to generate a structure with proper proportion, spatial position, and function. This communication must persist throughout the adult life to maintain the organ in light of environmental insults, accumulation of mutations, and during injury. New techniques such as single cell RNA-sequencing (scRNA-seq) allows for extraordinary views into the life of a single cell and how it interacts with neighboring cells and the environment. Using these techniques, we are able to recapitulate the diversity of cell types that make up skin and observe new cell types and cell-cell interactions previously unknown to exist. The goals of this research module will be to 1) explore clustering algorithms to better characterize cell types from scRNA-seq datasets, 2) define where extracellular signals originate in the skin and how they are coordinated to achieve the desired response, and 3) determine transition states between subpopulations of cells to predict cell fates. We will use these data to construct models on how specific cells respond to signaling events over time. These models will instruct how we experimentally interrogate cells at an organismal level through a range of genetic and molecular techniques to understand how cell-cell communication is vital for skin development and homeostasis.
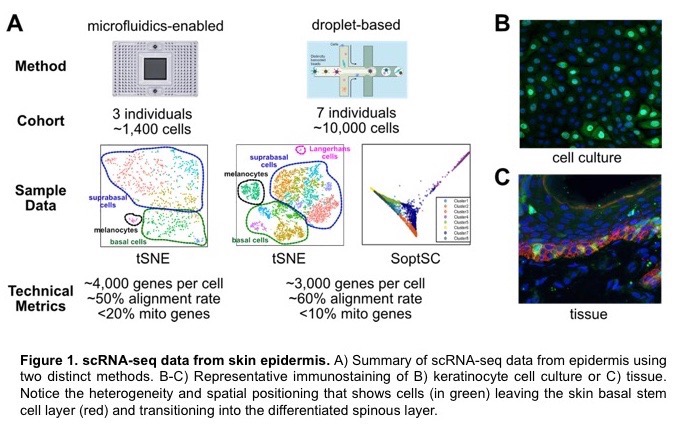
The Dai lab has been studying the molecular control of stem cell activity in the development, regeneration, and disease of mammalian epithelial tissues such as skin and mammary gland. Recently, we have used single-cell RNA-sequencing (scRNA-seq), a cutting-edge technology that enables the dissection of gene expression profiles of thousands of cells at once, and the associated bioinformatics methods to characterize the previously unknown cellular and molecular heterogeneities in normal tissues as well as under pathological conditions. My lab will offer opportunities for high school and undergraduate students to learn basic methodologies of analyzing scRNA-se data and beyond, to be involved in the development and application of new data analysis pipeline for defining signaling events and state transitions between different cell types within the tissues, and to be exposed to wet-laboratory techniques that help validate the findings and predictions generated by “dry” data analysis. Above all, I will work with the students to teach them how we formulate important questions of interest to biology and human health, and how we design specific experiments to address the questions at multiple levels using multidisciplinary approaches.
We have been using single cell RNA-seq to study differentiation of the mouse interfollicular epidermis. In these studies, we have identified Merkel cells — rare cells that have a specialized function in touch sensation as they are located at the ends of touch neurons. Merkel cells have features of neuronal cells despite being derived from epidermal cells. The development and differentiation trajectory of these cells are poorly understood. Our single cell RNA-seq datasets from embryonic and neonatal skin gives us a unique opportunity to gain new insights into Merkel cells. Specifically, 1) is there any heterogeneity of Merkel cells in the newborn epidermis? Are some Merkel cells still proliferating in the newborn epidermis? 2) Can we trace back the origin of Merkel cells to their earliest progenitors? 3) Can we identify new candidate regulatory factors in the differentiation trajectory? We will use an integrated experimental and computational approach to address these interesting questions.
DNA methylation is a type of epigenetic modification that is broadly associated with silencing of genes. Once established, cell-type-specific patterns of methylation are typically stable, but aberrant methylation is associated with disease and aging. In this project, we will be developing mathematical models of the enzymatic reactions controlling DNA methylation in order to address open questions about how DNA methylation patterns are established and maintained in vivo. For example, are there enzymatic mechanisms and/or structural features of DNA that can explain the long-range communication between individual nucleotides undergoing methylation, which has been observed in recent experiments? This project will build on previous studies that combine stochastic models with analysis of genomic data.
Solid tumors exhibit heterogeneity in metabolism that underlie adaptive strategies to challenging environmental growth parameters, e.g. nutrient deprivation and anoxia. A classical example is the Warburg Effect that describes the switch between oxidative phosphorylation and aerobic glycolysis. Evidence from solid tumor models, e.g. colon SW480 xenografts (Waterman, Lowengrub), implicates Wnt signaling as a contributing signaling mechanism for establishing tissue patterning for this metabolic switch.
Students will conduct metabolomic experiments with SW480 and SW480 dnLEF (dominant negative LEF) cell lines using a variety of high-end mass spectrometry instruments and bioinformatic metabolomic software tools. Global untargeted metabolomics will seek to identify potential biomarkers that are differentially expressed between the two cell lines. Students will apply statistical methods (PCA, o-PLSDA, clustering) to classify such component features and metabolite databases to identify selected biomarkers. Quantitative targeted metabolomic experiments will seek to validate candidate metabolites.
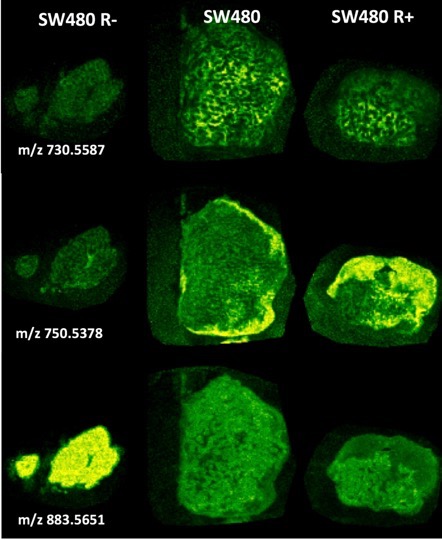
Students will also be able to interrogate and datamine an Imaging Mass Spectrometry (IMS) dataset obtained on a MALDI-IMS instrument of SW480 and SW480dnLEF xenografts for these specific biomarkers to determine any correlations to interesting metabolic domains and use mathematical transformations to derive parameters for any underlying patterns. MALDI imaging mass spectrometry is a new label free histology technique that collects mass spectra (of metabolites, lipids, peptides and proteins) pixel-by-pixel by raster scanning a laser across a matrix coated tissue specimen and is able to paint images by component masses rather than light. The figure shows some representative lipid metabolite distributions from this dataset that illustrates the types of heterogeneity observed in the tumors.
This project will complement and inform modeling from the project on Metabolic Symbiosis in Tumors (John Lowengrub).
Size control is one of the oldest and most fascinating problems in biology. The sizes of tissues and organs are specified with great precision, a fact we notice in the symmetry of bilateral structures (such as limbs), and the degree to which genetically identical individuals resemble each other. Not only do tissues and organs reach specific sizes, they do so in the face of cell killing or alterations to cell cycle kinetics, which suggests a feedback control mechanism. Work from UCI faculty on continually-renewing tissues has identified a general integral negative feedback strategy, whereby negative regulation of stem or progenitor cell renewal automatically achieves robust set-point control. Such feedback may be conveyed by diffusible growth factors — as we and others showed in the olfactory epithelium (OE), retina, and muscle — but a variety of molecules and mechanisms could act similarly. Regardless of mechanism, however, the ability of local feedback to control proliferation is subject to distance limitations: molecular and mechanical signals decay over characteristic length scales. The fact that such scales are often very short — on the order of 100 μm or less — raises questions about how local feedback could possibly control the sizes of tissues and organs that are three or four orders of magnitude larger.
The Controlled Shooting project exploits both mathematical modeling and experimental manipulation of genetically-engineered mice to discover how the sizes of tissues and organs are precisely specified in the face of developmental variability and external changes. The results of this study will provide both basic insights into fundamental processes of development, and specific insights into size control in the nervous system. The results will be of direct relevance to disease such as microcephaly. Size control is inherently a mathematical problem. This project will help reveal general principles for size control, and their connections with Control Theory.
An important new frontier in cardiovascular research and regenerative medicine is in generation of patient-specific cardiomyocytes (heart cells) from induced pluripotent stem cells. Mathematical and computational modeling can play a role in this area by helping to unravel the complex dynamics of gene regulatory networks governing cardiac development. In this project, stochastic mathematical models of cardiac gene networks will be developed, with a focus on understanding the link between molecular processes occurring during development and the efficiency of generating cells with desired properties. The model will be used to make experimentally-testable predictions about how various types of molecular perturbations affect phenotypic or functional characteristics of engineered cardiomyocytes.