
T Cells are immune cells responsible for surveilling the body for diseased cells – but sometimes they go awry, for example producing “cytokine storms” that have killed tens of thousands of humans in the past few months. To operate effectively, T Cells must push up against diseased cells and secrete toxic chemicals that are only received by the diseased cell, and not bystander cells. It has been hypothesized that the chemicals are released through a fluid-filled bubble called a secretory cleft. We previously used computational fluid dynamics to study the initial stages of T Cell contact [Liu, Read, Lowengrub, Allard 2019 PLoS Comp] applicable to CD4+ T Cells, which do not form secretory clefts. Here, we will use computational fluid dynamics to study the secretory cleft in CD8+ T Cells. How does the T Cell maintain a tight contact with the diseased cell, so no or few toxins leak out? The project involves the following. Tools: high-performance computing, Docker containers, bash and C++. Scientific topics: biophysics, fluid dynamics, immunology, cell biology.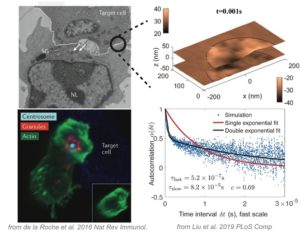
Psoriasis is a chronic autoimmune disease with a worldwide prevalence of around 2%, though prevalence varies based upon region. Our project focuses on the use of single cell RNA sequencing data from a mouse model of psoriasis that uses imiquimod treatment to induce psoriasiform inflammation. Using our scRNA-seq data, we can identify major differences in specific cell types at a transcriptomic level in response to imiquimod treatment. The initial results of this experiment have shown that the psoriatic model upregulates IRF7 and interferon sensitive gene expression in immune cells, and, to a lesser extent, keratinocytes. However, the impact of its application on the keratinocyte subpopulations has not been fully characterized. Using Seurat, we perform integration and clustering to identify conserved cell states in the treated and control keratinocytes, as well as note any differences in proportions of different cell states. Additionally, we will construct a differentiation trajectory that will trace from the basal populations previously identified, to terminally differentiated keratinocytes. We can then identify how modules of genes change over pseudotime in both the control and imiquimod treatment. Finally, we can work to identify regulators of keratinocyte gene expression, and determine which regulators are conserved and which differ between the treated and untreated epidermis.
Demographic models study human population dynamics by tracing individuals of different age-groups. On the other hand, epidemiological models are used to understand the spread of disease in populations of people. In this project, we will combine demographic and epidemiological models to understand disease spread in populations of individuals with a realistic age structure. We will use real-world data on the amount of contacts that people of different age groups have with each other, and identify which types of contacts (school, work, home) are the most important in the epidemic spread.
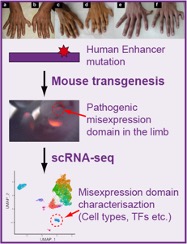
In the dawn of the modern era of genomics, we have made tremendous progress in understanding the effects of disease-causing gene mutations on the human organism. However, understanding the mechanism of disease-causing mutations located outside coding regions of genes remains a challenge. The vast majority of these mutations affect regulatory non-coding regions, called enhancers. Enhancers serve as platforms for binding regulatory proteins, which activate or silence the activity of genes in response to changing environmental and developmental stimuli. We will use limb as an established model system to study the mechanism of polydactyly-causing mutations in a critical limb enhancer of the Sonic hedgehog gene. Towards this goal, we will utilize single-cell gene activity profiling combined with mouse transgenesis to profile gene expression in transgenic mice that contain mutated enhancer. We will characterize the type of cells in which mutant enhancer causes increased gene activity by analyzing the global gene activity data. The single-cell resolution activity of mutant enhancers identified by this approach will provide insights into the functional properties of critical enhancers and the mechanism of their misregulation in human disease.
TA: Yutong Sha (Nie Lab)
A hallmark feature of many cancers, and an enduring puzzle of biology since its discovery in the 1950’s, is a phenomenon called the Warburg Effect. Here, cancerous cells gain access to oxygen, but then shift their metabolism to aerobic glycolysis, away from (oxygen-using) oxidative phosphorylation (OXPHOS) to favor glycolysis, despite the availability of sufficient levels of oxygen. This is especially intriguing since glycolysis is much less efficient than OXPHOS in producing energy. However, it is increasingly recognized that glycolysis is not a singular choice for all cells in a tumor; OXPHOS modes of metabolism may be dominant to some cells but not others. The proposed outcome of this heterogeneity is that cooperation between these two groups of cancer cells can maximize the delivery and consumption of nutrients and minimize the environmental stresses that are imposed on a tumor. This is known as metabolic symbiosis. For example, glycolytic cells produce lactate that can be used as fuel by cells using OXPHOS. Figure 1 shows metabolism under varying oxygen concentrations.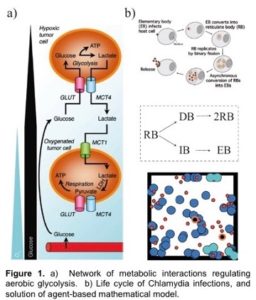
In this project, students will develop deterministic and stochastic population-based and individual-based mathematical models to determine conditions under which metabolic symbiosis is not just a manifestation of tumor heterogeneity, but is a fundamental aspect of tumor survival. That is, students will determine conditions and parameter ranges for which tumors that display metabolic symbiosis have an enhanced fitness compared to those in which there is metabolic homogeneity. The population-based modeling will use ordinary and stochastic differential equation models while the individual-based modeling will use CompuCell 3D, which is based on the Cellular Potts approach, as the primary modeling tool. We have extensive experience using these approaches. Model predictions will be compared to results in the experimental literature.
The mathematical challenge lies in developing a model that, on the one hand, contains all the relevant biological features needed to compare with experimental data, and, on the other, is simple enough to provide insight into the underlying mechanism of spatial heterogeneity.
In multi-cellular organisms, cells contain the same genetic code but specialize into many different tissue-specific phenotypes. This specialization is possible because of epigenetics—processes which act “above” (epi-) the genetic sequence to control gene expression. In this project, mathematical models will be used to study a particular type of epigenetic mark known as DNA methylation. We will be studying the stability of DNA methylation patterns using nonlinear dynamical systems models (patterns mean: which areas of the genome tend to be highly methylated versus demethylated in a particular cell type). DNA methylation patterns across the genome are generally very stable, and yet it is also known that changes in these patterns happen gradually with age and are implicated in cancer. Mathematical modeling will help us understand how various molecular processes that control DNA methylation patterns (informed by recent experimental findings) affect their long-term stability.
Biologists aren’t out of a job but robots can perform better when it comes to high-throughput discovering. Robots not only work faster than humans, but also avoid human errors in everyday experimental operation. Among all types of biological experiments, imaging using microscopes is one of the most complicated and low-throughput benchwork. Usually, the higher resolution the microscope offers, the lower throughput it has. Therefore, automation of the super-resolution microscopy is urgently needed. In this project, we will develop software to convert a manual super-resolution microscope to a fully automated robotic microscope. The software will control all motorized parts in the microscope and enable the microscope to optimize imaging conditions, and take images of cells and tissues with super-resolution and high throughput.
The MYC transcription factor regulates cell growth and proliferation, and its over-expression or deregulation can lead to many types of human cancer. Use various modeling and simulation techniques, we explore how Myc and related proteins form dimers with each other and walk along DNA, and how the local diffusional dynamics can possibly impact on the protein global search on the genome.
 scATAC-seq is a powerful approach for characterizing cell-type-specific regulatory landscapes. However, it is difficult to benchmark the performance of various scATAC-seq analysis techniques (such as clustering and deconvolution) without having a priori a known set of gold-standard cell types. To simulate scATAC-seq experiments with known cell-type labels, we introduce an efficient and scalable scATAC-seq simulation method (SCAN-ATAC-Sim) that down-samples bulk ATAC-seq data (e.g., from representative cell lines or tissues).Our protocol uses a consistent but tunable signal-to-noise ratio across cell types in a scATAC-seq simulation for integrating bulk experiments with different levels of background noise, and it independently samples twice without replacement to account for the diploid genome. Because it uses an efficient weighted reservoir sampling algorithm and is highly parallelizable with OpenMP, our implementation in C++ allows millions of cells to be simulated in less than an hour on a laptop computer.
scATAC-seq is a powerful approach for characterizing cell-type-specific regulatory landscapes. However, it is difficult to benchmark the performance of various scATAC-seq analysis techniques (such as clustering and deconvolution) without having a priori a known set of gold-standard cell types. To simulate scATAC-seq experiments with known cell-type labels, we introduce an efficient and scalable scATAC-seq simulation method (SCAN-ATAC-Sim) that down-samples bulk ATAC-seq data (e.g., from representative cell lines or tissues).Our protocol uses a consistent but tunable signal-to-noise ratio across cell types in a scATAC-seq simulation for integrating bulk experiments with different levels of background noise, and it independently samples twice without replacement to account for the diploid genome. Because it uses an efficient weighted reservoir sampling algorithm and is highly parallelizable with OpenMP, our implementation in C++ allows millions of cells to be simulated in less than an hour on a laptop computer.