
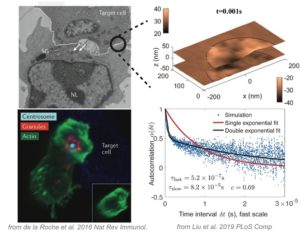
Hair loss is a problem that affects millions of people around the world. One promising treatment is to induce hair growth in waves, but this requires differentiating between different stages of the hair growth cycle on humans. We use a non-invasive skin imaging modality called OCT (pictured below) to find imaging parameters in order to make these hair cycle staging decisions. The goal of this project is to help process these 3D images and use machine learning to differentiate between the stages of hair growth. This would involve some classical image analysis as well as exposure to deep learning for image processing.
TAs: Sean Kuan (Plikus lab) and Eric Chang (Nie lab)
Noise in Genomic Sequencing (Elizabeth Read)
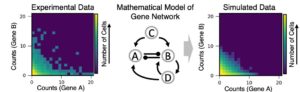
In recent years, new technology in genomic sequencing has enabled researchers to measure the expression of many thousands of genes simultaneously within individual cells. In a population of cells (extracted, for example, from animal or human tissue), these single-cell-level gene expression measurements appear very noisy—that is, they show big differences between individual cells. This project asks the question: How much of the noise in these data is useful noise? In other words, do cell distributions contain hidden information about underlying regulatory relationships between genes, and therefore about the gene networks that help determine an individual cell’s identity? Inferring such gene networks is an ongoing challenge for fundamental biological discovery and applied biotechnology. To address these questions, we will use mathematical modeling and simulation of small gene regulatory networks with defined characteristics. Noise properties of simulated networks will be compared to those of experimental data in order to determine the limits of what the noise properties can and can’t tell us about the underlying networks.
Mapping Electrostatic Impacts of Antiviral Drugs on Viral RNA Polymerase in SARS-CoV-2 (Jin Yu)
RNA-dependent RNA polymerase (RdRp) is a critical enzyme responsible for replication and transcription in RNA viruses such as the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) that causes current coronavirus disease 2019 (COVID-19) pandemic.
Investigating the mechanism of action of RdRp from SARS-CoV-2 (Cov-RdRp) offers promise for development of new antiviral therapeutics and for understanding the emergence of drug resistance. Our computational biophysics lab (https://sites.uci.edu/jinyulab/) employs molecular modeling and simulation techniques to investigate the structural dynamics underlying the Cov- RdRp function (Fig 1 left). For example, Remdesivir, a prodrug of a nucleotide analog, will be utilized to interrogate RdRp fidelity and better understand the basis for its antiviral activity (Fig 1). Our computation is currently supported by the COVID-19 HPC Consortium, using the world leading supercomputer Summit from the Oak Ridge National Lab.
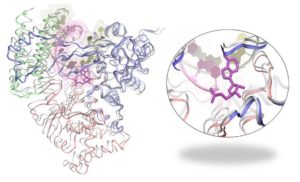
Fig 1: The viral RdRp protein enzyme from SARS-CoV-2 (left) and a close view of its active site docked with a nucleotide analog from the anti-viral drug compound Remdesivir
In this project, we focus on solving the Poisson-Boltzmann equation of the antiviral drug bound Cov-RdRp system, in order to map electrostatic impacts of various drug compounds on the Cov-RdRp protein. We will use software with the specialized Poisson-Boltzmann solver in combination with molecular graphics and visualization tool, molecular docking and force field optimization, and molecular dynamics simulation packages to accomplish the computational tasks. The key scientific question remains open and challenging for this project is, whether and how various potential drug molecules impact on the Cov-RdRp functioning electrostatically upon binding/incorporation.
See Dr. Yu’s project intro video here.