
Cells have the ability to control their rigidity — that is, the structural property of a material that allows it to withstand mechanical force — over orders of magnitude in minutes. This is an ability that material engineers aspire to recreate. It is also an ability that goes wrong in many disease in which cells are either too stiff (parasitic infection like malaria, some anemias) or too soft (some cancers and heart disease).
Actin, a key component of the cell cytoskeleton, is a filament-like protein which is the primary contributor to cell rigidity in mammalian cells. Recent work has shown that actin organizes into star-shaped patterns called “asters” that then crosslink into larger networks, yet little work has been done to relate these microstructures to larger-scale properties like strength and stiffness of a cell. In this project, we will model actin networks composed of various astral microstructures and explore how their mechanical properties may be varied through varying the properties of the asters. Previous modeling of polymer network strength computed network responses in the absence of thermal and biochemical activity, but at the cell scale such phenomena may significantly affect structural integrity. We will use a mechanical modeling platform Cytosim to simulate dynamic networks of actin filaments and extract their mechanical properties. We aim to demonstrate a clear dependence of shear modulus on the number of filaments per aster. In particular, we aim to identify optimal geometric properties of the asters which contribute to maximum network rigidity. This may suggest a mechanism by which cells may tune the physical properties of their actin networks, and may inform efforts to create synthetic metamaterials inspired by actin networks.
The team will use cell biophysics, mechanics (mechanical engineering), high-performance computing, Python, and discrete math.
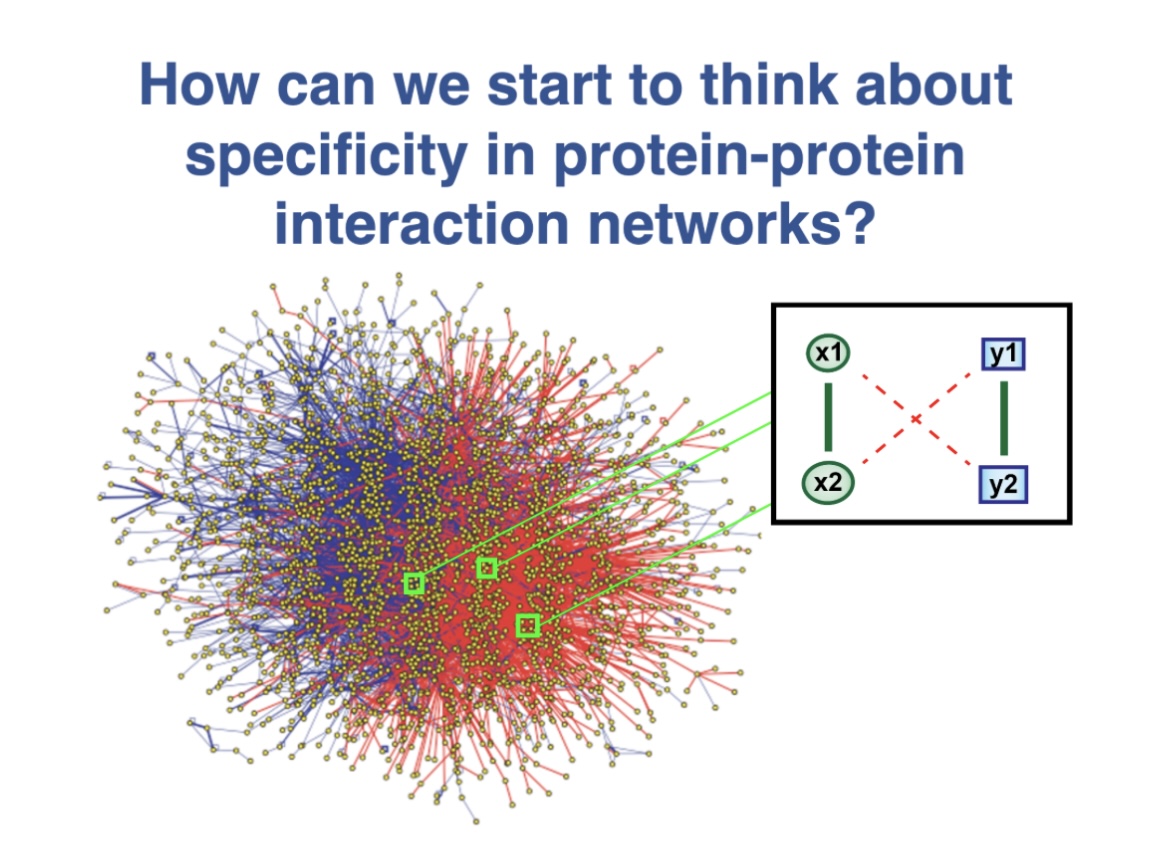
This MathBioU project concerns a new theoretical framework for the analysis of specificity in protein-protein interaction networks that I have been developing. Students will work using Mathematica software to help me further analyze this system.
Many studies that have shown that promiscuous protein-protein interactions exist, can have deleterious consequences for cells and organisms, and can impose constraints on evolutionary change. Indeed, as a consequence of the law of mass action, any two molecules will associate if their concentrations are high enough. For proteins that tend to be sticky, these “dangerous concentrations” can readily be reached in a cell, especially if the proteins are overexpressed. Moreover, other perturbations to protein-protein interaction networks, such as aneuploidy, viral attack, and even evolution, can also degrade specificity and efficiency, contributing to cancer, neurodegeneration, and other pathologies.
The project is similar in spirit to the published papers below, but considers protein-protein interaction networks rather than signal transduction networks:
Komarova, NL, Zou, X, Nie, Q and Bardwell, L (2005) A theoretical framework for specificity in cell signaling. Molecular Systems Biology, 1:2005.0023, doi:10.1038/msb4100031.
Bardwell, L, Zou, X, Nie, Q and Komarova, NL (2007) Mathematical models of specificity in cell signaling. Biophysical Journal, 92:3425-41.
Many cells in the body are surrounded by tissue, such as those made from familiar collagen. We are investigating how cancer cells can find each other and find other cell types by feeling vibrations traveling through the three-dimensional ‘spider web’ within a tissue. Participants will learn about a new imaging technique to detect such vibrations and work with PhD student Sarah Eldeen to develop and employ exciting equations that describe such vibrations, and importantly, use vibrations as a way of measuring forces within the web. An understanding of such force paths though the web, and how cells use molecules to detect them may unlock new strategies for fighting cancer.
Characterizing the metabolic signature of a cell is extremely important to understand dynamic processes such as the formation of cancer metastases. In our lab, we have developed a technique that utilizes hyperspectral fluorescence microscopy to visualize the metabolic response of multiple organelles (mitochondria, lysosomes, lipid droplets and the nucleus) to assess the cell’s metabolic state and how it is modified by the environment. Currently implemented algorithms, however, limit the applicability of the technology to a large number of cells (>10,000), as faster acquisition results in poorer image (and therefore analysis) quality. For this project, we will work on the implementation and fine-tuning of state-of-the-art denoising algorithms based on convolutional neural networks, as well as in the development of a novel algorithm for hyperspectral unmixing. The course will involve programming in Python and analysis of multidimensional microscopy datasets for the study of cancer metastasis.
Ultrasensitive dose responses allow biological systems to take decisions, in that these responses have either very large or very small outputs as a function of a continuously increasing input. How to create such all-or-none respones using only widely available tools from biochemistry is an important challenge when understanding signalling responses and cellular communication. We explore the question of composing multiple ultrasensitive responses as a way to increase the ultrasensitive behavior, as well as exploring a new mechanism of ultrasensitivity involving saturating functions.
Cancer tumorigenesis results from disturbances in regulatory control mechanisms to transform normal cells into a state of unregulated cell division and excessive growth. As tumor mass and cell number increase, cells continue to be subjected to selective pressures (low oxygen tension, changes in metabolic fuel sources and pH changes etc.) and evolve to adapt to these challenges. A tumor will contain many tumor cell variants and establish spatially distinct ecological niches. This variability can also make tumor cells differentially sensitive to therapeutic agents as metabolic pathways related to oxidative stress and drug metabolism are modulated. One key metabolic hallmark of many solid tumors is the Warburg effect where tumor cells switch from oxidative phosphorylation (TCA cycle) to aerobic glycolysis. Broad effects on many cellular metabolites are observed.
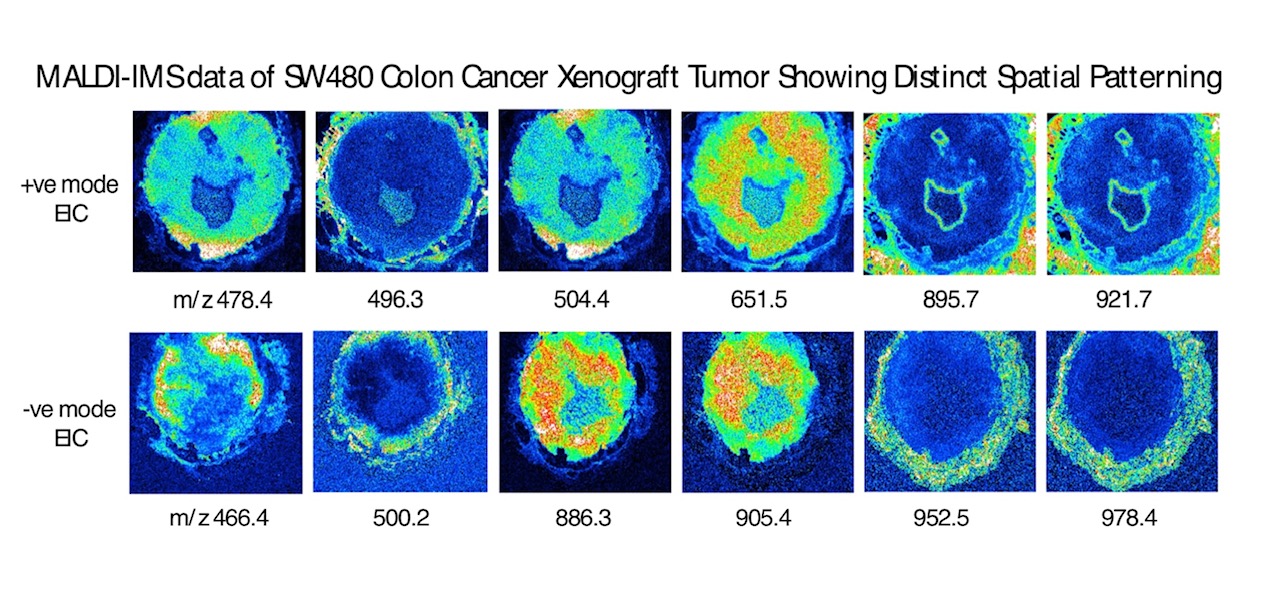
One approach to characterize these changes is with Imaging Mass Spectrometry (IMS) where histological sections of tumors can be analyzed for changes in metabolite, lipid and peptide components by mass spectrometry. These data can quantitate many individual molecular species while retaining their spatial information with the tumor.
Project goals are to collect and statistically analyze IMS data on colon tumor specimens and identify patterns of specific tumor metabolic markers. Data can be combined with other omics datasets (untargeted LC-MS metabolomics data) to generate hypotheses related to cancer progression.
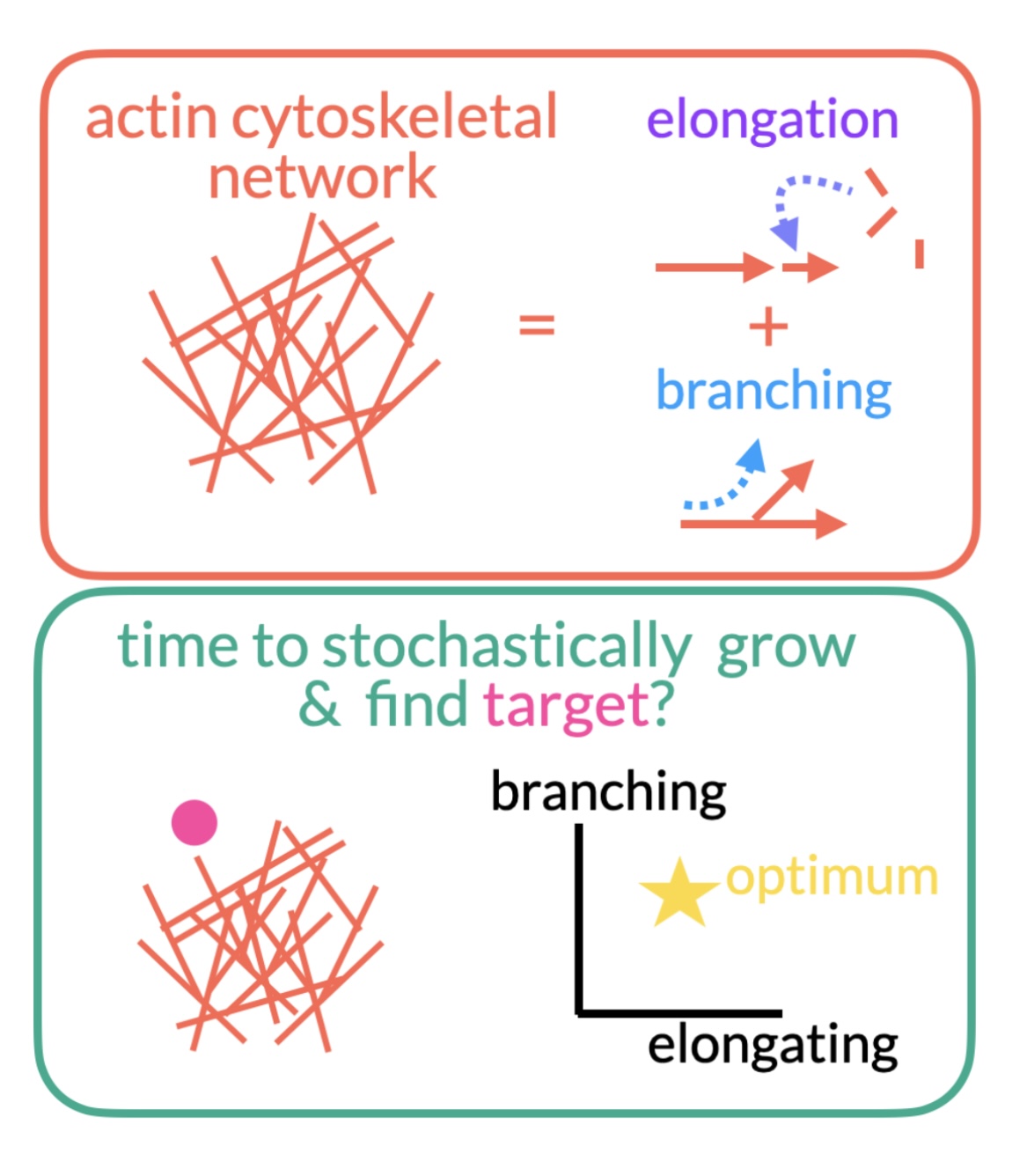
Cells rely on a network of cytoskeletal filaments called the actin cortex. The cortex is constantly evolving through a mixture of stochastic (random) elongation and branching. The relative frequency of each is constrained by cellular resources. How do these constraints affect the growth of the network? What balance of branching and elongation is optimal for finding a target in the shortest amount of time? Does this change for multiple targets? The project will primarily center around developing stochastic simulations of the growth of actin networks to explore these questions.
Adult tissue size and shape can be determined by multiple factors, including cellular proliferation, cellular volume expansion, and extracellular matrix (ECM) deposition. In the growth plate of long bones like the femur and the tibia, cartilage progenitor cells proliferate into organized “stacks” and differentiate as columns of cartilage cells, also known as chondrocytes. Mature chondrocytes expand their volume through a process known as hypertrophy, and eventually die via programmed cell death, leaving behind a columnar scaffold of ECM onto which bone cells will eventually grow. Other vital cartilage types, like those in the head, neck, and thorax, achieve their final shape through mechanisms that are not yet well understood. Here, we explore how specialized lipid-filled chondrocytes, known as lipochondrocytes, coordinate cellular proliferation and cell volume expansion to achieve precise tissue shape and size of the ear pinna, a nearly 2-dimensional sheet of tissue. Our studies show that the final shape and size of the ear pinna is determined after birth and in two steps: first, lipochondrocyte (LC) progenitors expand in number using as-of-yet undefined proliferative strategies; and second, they expand in volume via lipo-hypertrophy and self-organize into a large sheet of cells 2-3 cells thin. In combination, these two mechanisms achieve precise ear shape and size consistently across individuals. Here, we will try to find a model that can help us understand what mode of cell division (i.e., symmetric vsasymmetric) is used by LC progenitors to precisely reach the number of cells required to grow the pinna. Additionally, we will explore how lateral displacement by lipo-hypertrophy may optimally organize cells into a nearly 2-dimensional plate with a patterned topology.
DNA is more than just a string of letters (or nucleotides): in addition to this genetic code, the epigenetic code is crucial for determining which genes are expressed in a cell. Epigenetics involves a number of processes, from chemical modifications to molecular interactions to rearrangements of the 3D configuration of DNA. To make matters even more complicated, these processes are not constant in time, but dynamic. We will be carrying out statistical analyses of epigenomic data, along with mathematical modeling, to study the origins and implications of long-range correlations in epigenomic signals (that is, signals that are correlated even though they are many basepairs apart). We hope this project will help shed light on how different epigenetic processes work together to regulate gene expression.
Our lab studies the gene networks driving neuronal diversity during brainstem development to uncover why only specific subsets are differentially affected in a given neurologic disease. We first need to understand the normal transcriptomic differences between subsets of developing mouse motor neurons as a foundational model. The goal of this project is to utilize our existing temporal single-cell and spatial RNA-sequencing data to establish genetic fingerprints for various developing motor neuron subpopulations using pseudobulk differential gene expression analysis and then to map these subpopulations onto parallel spatial RNAseq datasets. Students will be introduced to single cell and spatial RNA-seq data analysis pipelines.
Early 2020 saw the start of the coronavirus disease 2019 (COVID-19)
pandemic, which is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). By January 26, 2020, two cases of novel coronavirus (2019-nCoV) have been reported in California. In December of 2020, CSUDH started registering and sharing the number of people that visited campus and were infected with COVID-19. On this report, they also included the places they visited and the days when this visit occurred. In this work we plan to use statistics and compartmental model/s (System/s of Ordinary Differential Equations) to study the CSUDH data. We would like to identify the rate of infection at each of the different stages of the epidemic and the different dynamics of campus (complete quarantine, partial quarantine, and no quarantine). Our aim is to understand the dynamics of COVID-19 on the CSUDH population. We expect that this work can serve for future epidemic occurrence and, if possible, to give a better understanding on which strategies implemented on campus were the most successful to minimize infections on campus.