Our lab uses physics to understand components inside cells. One such cellular component is the primary cilium, the “antenna” of the cell used to receive signals from other cells. It is used by human cells in many roles, including skin healing and brain structure. Defects in the cilium are connected to disease, including skin cancers and neurological conditions like schizophrenia, as studied by labs at UCI (one [Atwood Lab, Dev & Cell] that studies skin, and one [Alachkar Lab, Pharm Sci] that studies brain) show that certain cilia change length and molecular content during disease. How does that affect signaling? Since the circumstances are so different, we speculate that there is a mathematical reason. We will use a one-dimensional advection-reaction partial differential equation model to test these ideas. This model will involve a high-performance computing particle-based reaction-diffusion solvers, biophysics, and cell biology.

In order to communicate with each other and sense their environment, cells pull on their surroundings comprising rope-like collagen fibers that are in turn connected to other fibers and cells in a complex mesh. Participants in this project will work on analyzing images of collagen fiber networks to better understand how force travels through these fibers and allows cells to communicate with each other and their environment. Understanding this Cell-Extracellular Matrix interaction will provide insights into cell differentiation, vascularization, wound healing, and the onset of cancer; as well as help us develop better therapeutics, design interventions for various diseases, and create more effective biomaterials.
 Flower petals, tree branches and pineapple eyes have specific patterns that may optimize for functions such as proper spacing at different times during plant growth. It has been observed that the number of spirals in these patterns are often Fibonacci numbers, for reasons that are not well understood. In this project we will explore this pattern, and study how Fibonacci spirals may better fit optimization criteria than non-Fibonacci configurations. We will also study whether this pattern may appear from simple sets of criteria during plant growth, in order to determine the mechanism for pattern formation from first principles.
Flower petals, tree branches and pineapple eyes have specific patterns that may optimize for functions such as proper spacing at different times during plant growth. It has been observed that the number of spirals in these patterns are often Fibonacci numbers, for reasons that are not well understood. In this project we will explore this pattern, and study how Fibonacci spirals may better fit optimization criteria than non-Fibonacci configurations. We will also study whether this pattern may appear from simple sets of criteria during plant growth, in order to determine the mechanism for pattern formation from first principles.
Mathematical models are crucial to understand normal and abnormal biological processes. However, a model is only as good as its parameters. To determine the parameters, one needs to solve an inverse problem in which the parameters are chosen so that the model solution provides a “best” fit to data. In this project, we will develop methods for solving inverse problems in biological systems using deep-learning methods such as physically-inspired, neural networks. In this approach, both the model and parameters are embedded in the loss function and using a constrained optimization approach, the weights of the neural network and the model parameters can be determined simultaneously. We will start with simple model problems and aim to build up to solve equations modeling the growth of glioblastoma, a deadly brain tumor, using magnetic resonance images (MRI) from clinical patients to constrain the mathematical models and determine the parameters in the equations. We will mainly use python as the computing language.
It is now possible to take images of the individual mRNA molecules inside the nucleus of a cell. The spatial distribution and counts of these molecules encode the notion of “gene expression”, e.g., which of your genes are “activated”. However, decoding this data into biologically meaningful results is challenging because the processes underlying gene expression are very stochastic (random) and the nucleus is a very heterogeneous, crowded place. In this project, we will combine spatial stochastic simulations with simulation-based statistical inference (approximate Bayesian computation) to (1) identify the underlying processes of gene expression and (2) disentangle the spatial heterogeneity of the cell nucleus.
In this project, students will use computational tools to investigate the ways in which cells communicate by sending each other signals. By releasing a signaling protein (called ligands) that can be detected by receptors on other nearby cells, cells can communicate with each other and organize collective behavior. Given a piece of biological tissue, we can measure what genes are being expressed by each cell, telling us what ligands and/or receptors it is producing. By applying computational libraries such as CellChat and exFINDER to such datasets, we can then infer what kinds of communication are occurring in that tissue.
Given two different samples of tissue (such as one healthy sample and one diseased sample), we would like to be able to answer questions such as: What types of signaling are stronger or weaker in each sample? How big is the difference, and is it statistically significant? What does that tell us about what might be causing the disease, or affected by it? By investigating datasets on diseases like intestinal inflammation, Alzheimer’s disease, and cancer tumors, we will study how to combine existing tools with new ideas in order to answer these questions. Students will learn standard computational analysis pipelines for single-cell genomics data using R and/or Python, explore the data themselves, and then experience the process of developing and testing new methods in computational biology.
In multi-cellular organisms, cells contain the same genetic code but specialize into many different tissue-specific phenotypes. This specialization is possible because of epigenetics—processes which act “above” (epi-) the genetic sequence to control gene expression. In this project, stochastic mathematical models will be used to study a particular type of epigenetic mark known as DNA methylation. Mathematical modeling helps us understand how enzymatic processes and molecular features affect the stability of epigenetic marks. This in turn can help guide the development of new therapies. One current challenge lies in developing methods to fit mathematical models to real-world data, when modeling is computationally costly. To this end, we will be comparing the accuracy and efficiency of two different mathematical frameworks: a more detailed, stochastic simulation of DNA methylation dynamics, with a more coarse-grained numerical approach.
Our lab studies the gene networks driving neuronal diversity during brainstem development to uncover why only specific subsets are differentially affected in a given neurologic disease. We first need to understand the normal transcriptomic (gene expression) differences between subsets of developing mouse motor neurons as a foundational model. The goal of this project is to utilize our existing temporal single-cell and spatial RNA-sequencing data (RNA-seq) to establish the sequence of gene expression changes from dividing progenitor cells to mature motor neurons during brainstem development. Motor neurons form over several days and hence, at a particular time in development, motor neurons will exist in different cell states (i.e. closer to dividing progenitor cell vs. closer to mature motor neuron). One can line these cells up based on gene expression changes to generate a “pseudotemporal trajectory” of the cells. Using these models, one can uncover the order of gene expression changes in development and search for new genes involved in these processes. Students will be introduced to single cell RNA-seq data analysis pipelines and toolsets designed to generate these pseudotemporal trajectories.
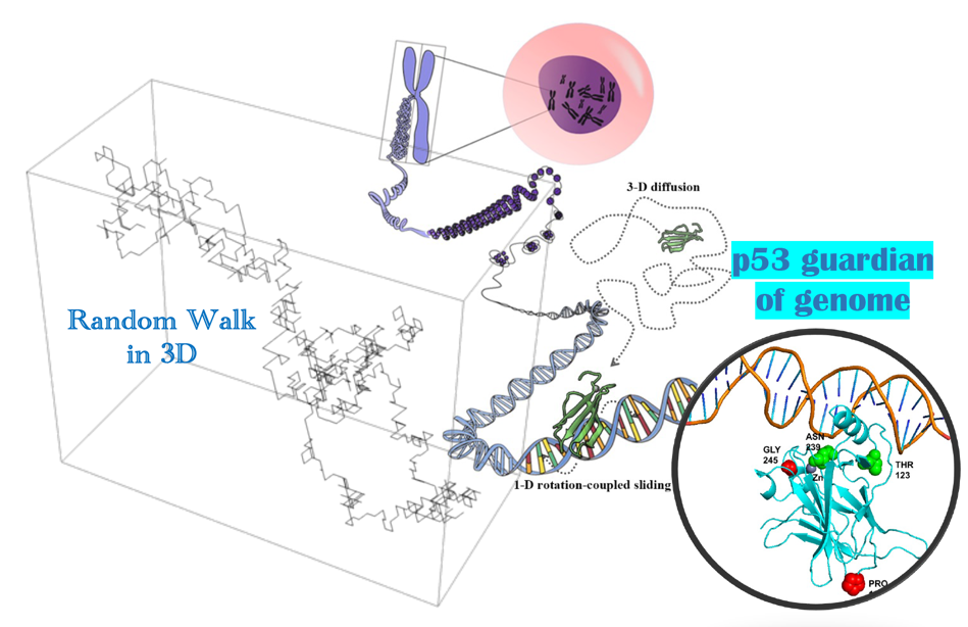
In this project, we start from generating random walk via stochastic methods, to figuring out the transcription factor (TF) target search on genome in a quantitative model, and then to deciphering mechanisms of the TF p53 protein mutants leading to human cancer, e.g., by employing a “computational microscope”.